
Sickle
Cell Anemia and Genetics: Background Information
Background
information to accompany the labs: Allele Frequencies and Sickle Cell Anemia
Lab and Sickle Cell Anemia: Diagnosis Using Restriction Analysis of DNA
Genetics
of Sickle Cell Anemia
Sickle cell anemia was the first genetic disease to be characterized at the molecular level. The mutation responsible for sickle cell anemia is small—just ONE nucleotide of DNA out of the three billion in each human cell. Yet it is enough to change the chemical properties of hemoglobin, the iron and protein complex that carries oxygen within red blood cells.
There are approximately 280 million hemoglobin molecules in each red blood cell (RBC). The protein portion of hemoglobin consists of four globin subunits: two alpha and two beta. These two types of subunits are encoded by the alpha and beta globin genes, respectively. While the binding of oxygen actually occurs at the iron sites, all four globin chains must work together in order for the process to function well.
Sickle
cell anemia, also known as sickle cell disease, is caused by a point mutation
in the beta globin gene. As a result of this
mutation, valine (a non-polar amino acid) is inserted
into the beta globin chain instead of glutamic acid (an electrically charged amino acid). The
mutation causes the RBCs
to become stiff and sometimes sickle-shaped when they release their load of oxygen. The sickle cell mutation produces a
“sticky” patch on the surface of the beta chains when they are not complexed with oxygen.
Because other molecules of sickle cell hemoglobin also develop the sticky patch, they adhere to each other and polymerize
into long fibers that distort the RBC into a sickle
shape.
The sickled cells tend to get stuck in narrow blood vessels, blocking the flow of blood. As a result, those with the disease suffer painful “crises” in their joints and bones. They may also suffer strokes, blindness, or damage to the lungs, kidneys, or heart. They must often be hospitalized for blood transfusions and are at risk for a life-threatening complication called acute chest syndrome. Although many sufferers of sickle cell disease die before the age of 20, modern medical treatments can sometimes prolong these individuals’ lives into their 40s and 50s.
There are two beta globin alleles important for the inheritance of sickle cell anemia: A and S. Individuals with two normal A alleles (AA) have normal hemoglobin, and therefore normal RBCs. Those with two mutant S alleles (SS) develop sickle cell anemia. Those who are heterozygous for the sickle cell allele (AS) produce both normal and abnormal hemoglobin. Heterozygous individuals are usually healthy, but they may suffer some symptoms of sickle cell anemia under conditions of low blood oxygen, such as high elevation. Heterozygous (AS) individuals are said to be “carriers” of the sickle cell trait. Because both forms of hemoglobin are made in heterozygotes, the A and S alleles are codominant.
About 2.5 million African-Americans (1 in 12) are carriers (AS) of the sickle cell trait. People who are carriers may not even be aware that they are carrying the S allele!
Sickle
Cell Anemia and Malaria
In
the
The answer is related to another potentially fatal disease, malaria. Malaria is characterized by chills and fever, vomiting, and severe headaches. Anemia and death may result. Malaria is caused by a protozoan parasite (Plasmodium) that is transmitted to humans by the Anopheles mosquito. When malarial parasites invade the bloodstream, the red cells that contain defective hemoglobin become sickled and die, trapping the parasites inside them and reducing infection.
Compared to AS heterozygotes, people with the AA genotype (normal hemoglobin) have a greater risk of dying from malaria. Death of AA homozygotes results in removal of A alleles from the gene pool. Individuals with the AS genotype do not develop sickle cell anemia and have less chance of contracting malaria. They are able to survive and reproduce in malaria-infected regions. Therefore, BOTH the A and S alleles of these people remain in the population. SS homozygotes have sickle cell anemia, which usually results in early death. In this way, S alleles are removed from the gene pool.
In a region where malaria is prevalent, the S allele confers a survival advantage on people who have one copy of the allele, and the otherwise harmful S allele is therefore maintained in the population at a relatively high frequency. This phenomenon will be examined in the Allele Frequencies and Sickle Cell Anemia Lab, which relates the change in allele frequency in a population to evolution.
The
frequency of the S allele in malaria-infected regions of
Sickle
Cell Anemia and Current Research
The oxygen requirements of a fetus differ from those of an adult, and so perhaps not
surprisingly, prenatal blood contains a special hemoglobin. Fetal hemoglobin contains two gamma globin polypeptide chains instead of two adult beta chains. After birth, the genes encoding gamma globin switch off, and the ones encoding beta globin switch on. Understanding how this genetic switch works could allow researchers to understand much about the control of genes in general and sickle cell anemia in particular.
•
Some infants whose mothers suffered from diabetes during pregnancy have
unusually high concentrations of the biochemical butyrate in their blood
plasma. When butyrate is given to patients with sickle cell
anemia, the fetal hemoglobin production increases significantly. Perhaps
butyrate or other chemicals that stimulate fetal hemoglobin production could be
used to treat sickle cell anemia.
• Mice that have been genetically engineered to contain a defective human beta globin gene have symptoms typical of sickle cell anemia, making them an ideal model for laboratory experimentation. In 2000, these mice were mated to another transgenic mouse line expressing human fetal hemoglobin. When compared to their sickle cell parents, the offspring had greatly reduced numbers of abnormal and sickled RBCs, increased numbers of RBCs overall (reduced anemia), and longer lifespans. These experiments established that only 9-16% of hemoglobin need be the fetal type in order to ameliorate the sickle cell symptoms, and are an important first step in a gene therapy solution to sickle cell disease.
Sickle
Cell Anemia: Translation Practice Worksheet
After
reading Sickle Cell Anemia and Genetics Background Information, answer the
following questions.
Sickle Cell
at the Molecular Level
In sickle cell anemia, there is a mutation in the gene that encodes the beta chain of hemoglobin. Within this gene (located on Chromosome 11), ONE BASE in the DNA is replaced with another base, and this mutation causes the normal amino acid #6 to be replaced by another amino acid.
1.
Making a
The sequence below is the first part of the DNA sequence for the â chain of normal hemoglobin. Fill in the complementary DNA strand using the base-pairing rules for making DNA (A pairs with T, C pairs with G).
DNA: GTG
CAC CTG ACT
CCT GAG GAG
DNA:_________________________________________
Now make the messenger RNA from the new, complementary strand of DNA that you just wrote down. Use the RNA base-pairing rules (same as DNA but use U instead of T).
mRNA:_______________________________________
Now, using the Genetic Code chart in your textbook, translate this mRNA into a sequence of amino acids.
Amino
Acids:____________________________________________________________
2.
Making Sickle Cell Hemoglobin
In sickle cell anemia, there is a mutation at the seventeenth nucleotide of DNA in this gene; the nucleotide is changed from A to T. Fill in the complementary DNA strand, mRNA, and amino acid sequence in the hemoglobin protein.
DNA: GTG
CAC CTG ACT
CCT GTG GAG
DNA:_________________________________________
mRNA:_______________________________________
Amino
Acids:__________________________________________________________
3.
The Effect of Changing One Amino Acid
You can see that in normal hemoglobin, amino acid #6 is glutamic acid (Glu) and in sickle cell hemoglobin, amino acid #6 is valine (Val). Observe the two structural formulas for these amino acids:

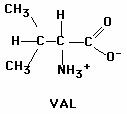
Describe which amino acid is polar and which one is nonpolar. How can you tell which is
which?
Although the altered beta globin has only one amino acid changed out of the total of 146, it’s acrucial amino acid. When this new amino acid is at position #6 instead of the correct amino acid, the overall hemoglobin â chain becomes more hydrophobic. As a result, when the hemoglobin chains fold into their 3-dimensional shape and assemble together, the resulting molecules tend to STICK TOGETHER, forming long chains of hemoglobin.
This altered hemoglobin deforms the normally rounded cell into the sickle shape. These red blood cells are destroyed at an increased rate, causing anemia. They are also prone to becoming stuck in capillaries, causing pain, organ damage, and often premature death.
Summary
1. How does sickle cell hemoglobin differ from normal hemoglobin at the primary level of protein structure (order of amino acids)?
2. How does sickle cell hemoglobin differ from normal hemoglobin at the fourth level of
protein structure (the sum of all the folded protein chains)?
3.
What is the effect on the red cell containing this altered hemoglobin?
Allele
Frequencies and Sickle Cell Anemia Lab
Student
Instructions
Objective: To observe how selective forces can change allele frequencies in a population and cause evolution to occur.
Background:
Read the background information provided in the handout, Sickle Cell
Anemia and Genetics: Background Information.
Introduction: Allele frequency refers to how often an allele occurs in a population. Allele frequencies can change in a population over time, depending on the ‘selective forces’ shaping that population. Predation, food availability, and disease are all examples of selective forces.
Evolution occurs when allele frequencies change in
a population!
In
this activity, red and white beans are used to represent two alleles of beta globin. The RED beans represent gametes carrying the beta globin A allele, and the WHITE
beans represent gametes carrying the beta globin
S allele. The Gene Pool exists in a region of
Materials:
75 red beans, 25 white beans, 5 containers (e.g. paper cups)
Hypothesis/Prediction:
What do you think will happen to the frequencies of the A and S alleles as a result of the
presence of malaria? (Will the frequency of A increase or decrease? What about S?) Formulate a hypothesis and corresponding prediction. Be sure to explain your reasoning.
Procedure:
1. Together with your lab partner, obtain five containers and label them as follows:
1) AA 2) AS 3) SS 4) Non-surviving alleles 5) Gene Pool
2. Place the 75 red and 25 white beans in the Gene Pool container and mix the beans up.
3. Simulate fertilization by PICKING OUT two ‘alleles’ (beans) WITHOUT LOOKING.
4. For every two beans that are chosen from the gene pool, another person will FLIP A COIN to determine whether that individual is infected with malaria.
5. Using the table below, the coin flipper tells the bean picker in which containers to put the beans.
Genotype
Phenotype Malaria (Heads) Not Infected (Tails)
|
Genotype |
Phenotype |
Malaria (Heads) |
Not Infected
(Tails) |
|
A A (Red-Red) |
No sickle cell disease. Malaria susceptibility. |
Die: place in Non-surviving |
Live: place in AA |
|
A S (Red/White). |
No sickle cell disease. Malaria resistance. |
Live: place in AS |
Live: place in AS |
|
S S (White/White) |
Sickle cell disease. |
Die: place in Non-surviving |
Live for a brief time: place in SS |
6. Repeat steps 3–5 until all the beans in the Gene Pool are used up.
7. At the end of the round, COUNT the number of individual red beans (A alleles) and white beans (S alleles) in the containers labeled AA and AS. These individuals survive to
reproduce. RECORD those numbers in the F1 TOTAL SURVIVING ALLELES table. Put them in the gene pool afterwards.
8. Because SS individuals do not survive to reproduce, move all beans from the SS alleles
container into the Non-surviving alleles container.
STOP AFTER ONE GENERATION.
CHECK WITH YOUR TEACHER BEFORE GOING ON!
9. Repeat the procedure for the F2 generation. Record your results in the F2 TOTAL
SURVIVING ALLELES table.
Data
Sheet for Allele Frequencies and Sickle Cell Anemia Lab
F1 TOTAL SURVIVING ALLELES: (very important to record)
|
Number of A (RED) alleles surviving (Count out of AA
and AS containers) |
|
|
Number
of S (WHITE) allele surviving (Count out of AS container) |
|
Put the survivors in the gene pool and create the next generation.
F2 TOTAL SURVIVING ALLELES: (very important to record)
|
Number of A (RED) alleles surviving (Count out of AA
and AS containers) |
|
|
Number
of S (WHITE) allele surviving (Count out of AS container) |
|
Class
Results
On the class overhead, record your number of A alleles surviving for the next generation and number of S alleles surviving from both the F1 TOTAL SURVIVING ALLELES and F2 TOTAL SURVIVING ALLELES tables. Then record the class totals below and calculate the frequencies using the formula below.
Using the formulas below, calculate the % allele frequency for each allele in each generation:
Total A x 100 = % Allele A Total S x 100 = % Allele S
Total A+S Total A+S
Class
Results Table
|
|
Parents |
F1 |
F2 |
|||
|
|
A |
S |
A |
S |
A |
S |
|
Class Total |
|
|
|
|
|
|
|
Allele Frequency |
|
|
|
|
|
|
Allele
Frequencies and Sickle Cell Anemia Lab Questions
1. What do the red and white beans represent in this simulation? What does the coin represent? (See background information.)
2. What do you think “allele frequency” means? How are allele frequencies related to
evolution? (See background information.)
3. What are the “selective forces” in this simulation (the forces changing the allele
frequencies)?
4. What was the general trend you observed for Allele A over the three generations (did it
increase or decrease)? What was the general trend for Allele S over time? Was your
hypothesis supported?
5. Since few people with sickle cell anemia (SS) are likely to survive to have children of their own, why hasn’t the mutant allele (S) been eliminated? (Hint: what is the benefit of keeping it in the population?)
6.
Why is the frequency of the sickle cell allele so much lower in the
7.
Scientists are working on a vaccine against malaria. What impact might the
vaccine have in the long run on the frequency of the sickle cell allele in
Sickle
Cell Anemia: Diagnosis Using Simulated Restriction
Analysis of DNA.
PreLab
Background,
Sickle Cell Anemia: Read the background information provided in the
handout, Sickle Cell Anemia and Genetics: Background Information.
Background,
DNA Restriction Analysis as a Diagnostic Tool:
DNA
obviously differs from one individual to another. However, some areas of DNA
contain quite a bit of sequence variation due to point mutations, deletions,
insertions, and repetitions. These areas are often referred to as polymorphic
regions (or “many-form places”). Alleles of a gene are a familiar example of a
polymorphism, for example, the A and S alleles of the beta globin gene. (Let A=allele for normal hemoglobin and
S=allele for sickle hemoglobin)
. When human DNA is digested with a particular restriction
enzyme, a polymorphic region yields fragments of different sizes, called RFLPs (pronounced “riflips”,
meaning “restriction fragment length polymorphisms” -whew!). The fragments are
separated by gel electrophoresis. The
patterns resulting on the gel can be used to identify criminals or settle
paternity cases. They can also be used in captive breeding programs of
endangered species (cheetahs and
PreLab Questions: Answer the following questions
1. What are RFLPs and how are they used?
2. How can a restriction enzyme help identify carriers of sickle cell anemia?
3. If
a person has sickle cell anemia and his or her beta globin
DNA is cut with Mst
II, will the fragments be longer or shorter than those from an individual
without the disease? Explain!
Sickle
Cell Disease Diagnosis Lab
Student
Instructions and Questions
Objective: To simulate the diagnosis of sickle cell anemia with DNA restriction analysis.
Background: The “DNA” you will receive has, in this simulation, already been “cut” by the Mst II restriction enzyme. You will separate the resulting fragments of DNA by gel
electrophoresis in order to diagnose the genotypes of all members of a family (mother, father, teenager, and fetus). Known samples will also be run for comparison. DNA and dyes are ‘charged’ molecules that can be separated by gel electrophoresis. The dyes
we will use are charged in solution, just as DNA is. They will therefore move from the BLACK cathode (- end) to the RED anode (+ end). (Remember, negatively charged molecules such as DNA “run towards the red.”)
Procedure:
1. Receive the seven “DNA” samples from your teacher. The tubes are coded in the following manner:
Mother M
Father F
Teenager T
Fetus O
Known
Known Carrier C
Known Sickle Cell Patient S
2. Using a micropipettor set to 8 ml, using key below, load each well in the gel with the correct samples. Take turns loading with others in your group.
3. If necessary, add more 1X TAE Buffer to the gel box so that the gel is adequately covered. (The buffer should cover the gel by about 1-2 mm.) Connect the electrodes to the gel box and to the power supply (red to red, black to black).
4. Turn on the power supply. Run the gel for at least 10 minutes. 5. Turn off the power supply, unplug the electrodes. View the completed gel run. Color the pattern observed into your RESULTS drawing.
1 2 3
4 5 6 7
__
__ __ __
__ __ __
 Key
Key
1 = Mother
2 = Father
3 = Teenager
4 = Fetus
5 = Known
6 = Known Carrier
7 = Known Sickle Cell
Patient
Analysis:
1. Intrepret the results of the tests:
• Which family members have the sickle cell genotype (SS)?
• Which family members have the carrier genotype (AS)?
• Which family members have the normal genotype (AA)?
2. Draw a pedigree showing inheritance of sickle cell anemia in the family you analyzed.
3.
Make a
4. Should genetic changes that cause hereditary problems be diagnosed before birth? (For your information, scientists estimate that each of us has at least six lethal recessive genes!)
5.
What are some possible sources of error in this lab?